这操作有点东西 (2)
前言
拖更好久的使用三代全长转录组注释基因组的一些思路,又结合一些其他测试的软件使用情况,做一个基因组注释的个人使用流程汇总。
首先通俗解释一下基因组注释:我们对一个物种进行基因组测序,组装得到的初步基因组序列是一本由“ATGC”组成的“天书”。我们需要进一步对这本“天书”进行“注解”,定义哪一页哪一行具体代表什么含义,也就是搜寻并定义基因组原件,得到其在基因组上的位置、序列、结构、功能等信息。
一般主要步骤为:重复序列鉴定 ==> 基于 de novo ,RNA 和 homology 方法进行基因结构预测 ==> 基因功能注释。
当然还有一些其他注释环节,比如非编码RNA 注释,启动子预测,假基因预测等,本文内容暂不涉及。
重复序列注释
集合重复序列注释的 pipeline 在 github 上有不少,这里提两个:
EDTA:https://github.com/oushujun/EDTA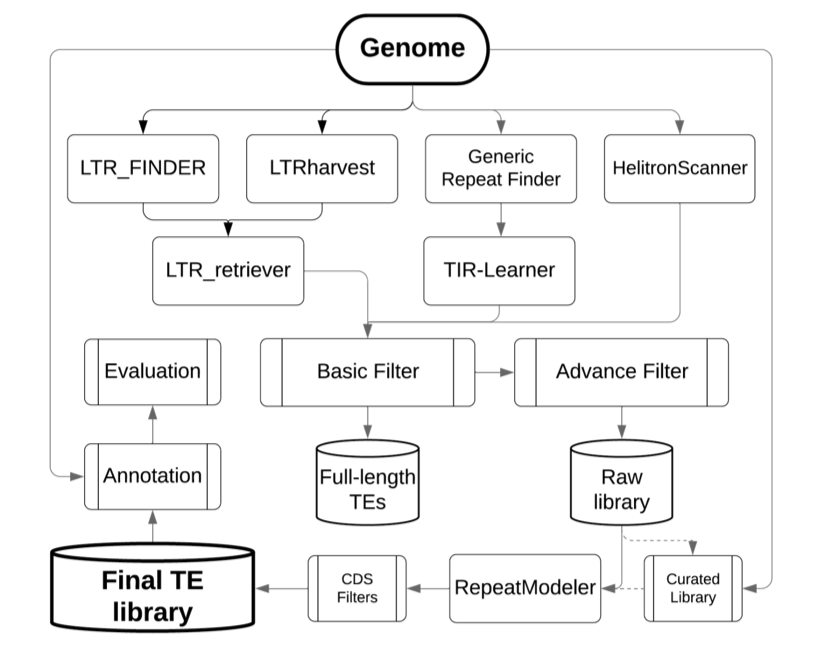
EarlGrey:https://github.com/TobyBaril/EarlGrey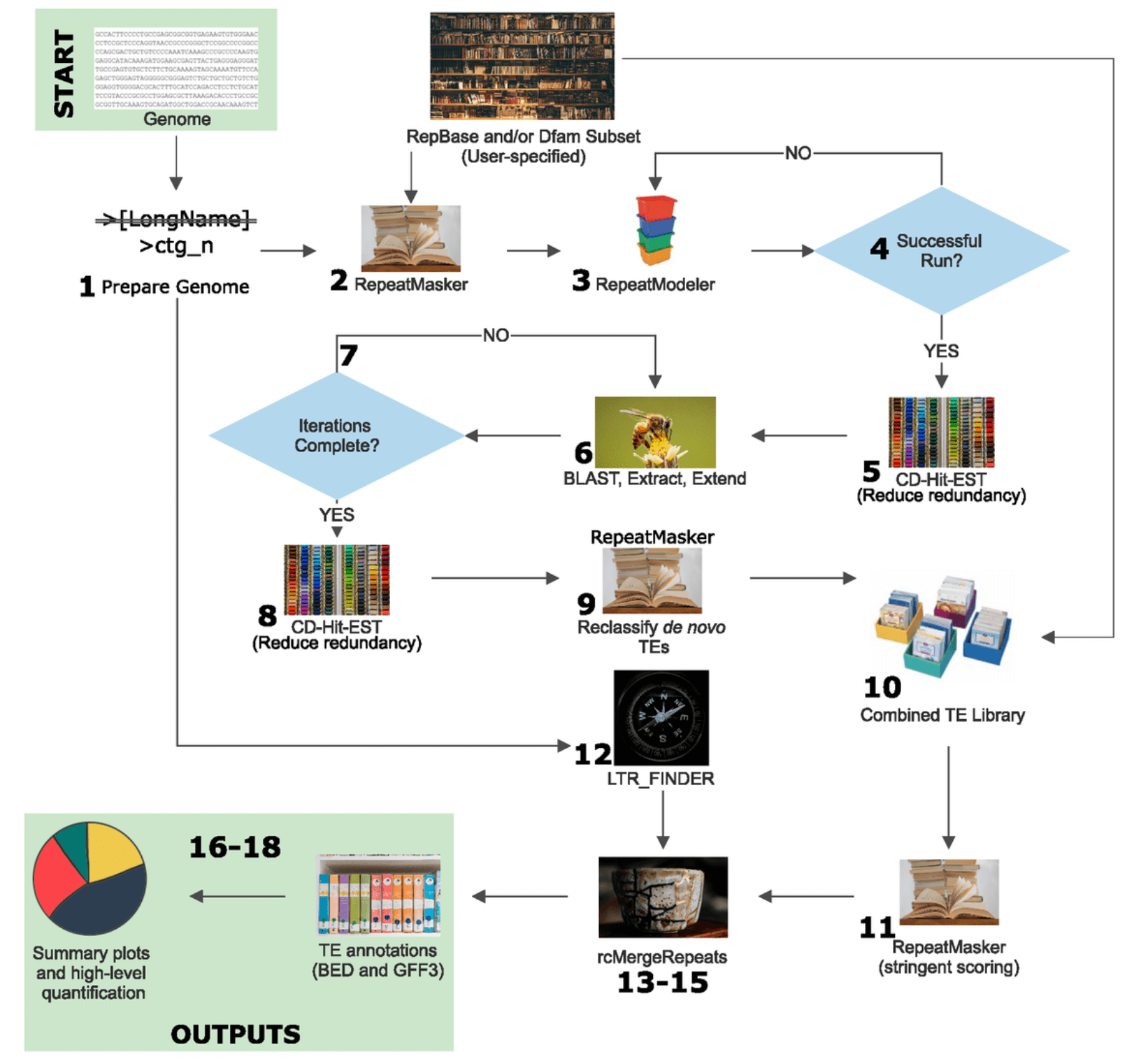
正好有个朋友在公众号分享了这两个软件测试后感受,见其公众号文章 基因组重复序列注释EDTA和EarlGrey问题一二。测试完两个软件后,他还是选择走了公司用的重复序列流程注释路子。可见公司的商业流程历经完善,满足客户需求,还是有一定道理的。
和他交流过程中,总结出几个问题和想法:
- 不同软件对于不同 class 的 transposable element (TE) 有一定偏好性;
- 动物和植物的重复序列有一定区别,有些软件适用性也与库有关,选好各自适用的软件;
- 许多 Unknown 分类的 TE。可以使用 DeepTE 或其他分类软件对 TE 再分类,之后重新对结果统计。
- 由于 TE 的嵌套,最后注释结果 TE 有很多 overlap。去冗余的话,可以试试 collapse_RM_annotation.py。RepeatMasker 自己流程里的 RM2Bed.py 也能部分实现冗余统计的功能。还有这位国外大神写的处理 RepeatMasker 结果的脚本也很实用 Parsing-RepeatMasker-Outputs
基于 RNA 数据注释
无论是 二代RNA-seq 数据,还是三代 full-length transcriptome 数据注释的基本思路大都是将转录组数据比对到基因组上,组装转录本序列,预测基因 ORF 结构,坐标位置整合基因组上。
RNA-seq
注释思路如下:
- 尽量选择更多多样性的 RNA 数据(不同组织,不同时期),使用 hisat2 比对得到 bam 文件;
- 组装转录本序列有两条路子:
- stringtie 软件 组装转录本序列;
- trinity 软件 根据 bam 文件,进行有参的方式组装转录本序列;
- 对转录本序列使用 TransDecoder 预测 ORF;
- 使用 TransDecoder 的 cdna_alignment_orf_to_genome_orf.pl 脚本,整合最终注释 gff 文件。
full-length transcriptome
注释思路如下:
- 使用 GMAP 软件将组装好的全长转录本序列比对到基因组上,结果输出为 gff 格式;
- gff 过滤。
- 因为GMAP 这样的输出结果中会有很多短于 3 碱基长度的 exon/CDS 结果,我觉得不太可靠,一般过滤短于 9 碱基长度的 exon;
- 去除一些存在 overlap 的冗余信息;
- 使用 gffread 软件 -N 参数去除具有非规范剪接位点 (i.e. not GT-AG, GC-AG or AT-AC) 的mRNA 信息。
PS:
- 其实 RNA-seq 和 full-length transcriptome 注释方法有些步骤都能交叉使用;
- full-length transcriptome 组装的全长转录本序列可以先 cd-hit 去冗余后,再比对到基因组上,我一般使用的参数:
1
cd-hit-est -i <input> -o <output> -c 0.99 -T 6 -G 0 -aL 0.90 -AL 100 -aS 0.99 -AS 30
- 使用Pacbio 的 Iso-Seq 组装结果,我也会将 singletons 序列用于注释。
homology 同源注释方法
注释思路如下:
- 选择 2-3 个近源物种的各自全部蛋白序列,使用 blat 比对后,再使用 genewise 预测。这个方法比较慢,也比较耗比对资源。
- 李恒大神的 miniprot 推荐可以试试,一个字——“快”,准确性也较高。因为最后都会用其他软件将这些结果整合到一起,也不用过于追求完美准确性。

PS:
这些结果其实也都可以用 gffread 软件 -N 参数过滤一遍。
de novo 注释
主要就推荐 AUGUSTUS 软件
注释思路如下:
- 根据 AUGUSTUS 训练集选择标准,从 RNA 数据组装的转录本序列中选择基因序列进行训练。一些步骤可以参考这个推文:AUGUSTUS模型训练。
- 训练完,基于模型从头预测就完事儿。
整合 de novo ,RNA 和 homology 注释结果
EvidenceModeler(EVM),GLEAN,maker 都能用来整合上面不同数据注释结果。
对于整合结果处理:
- 整合后,基因数目很少。优先从 RNA 数据注释结果中补充。
- 整合后,基因数目异常多。
- 基因结构不完整的可以考虑去除;
- 结合证据来源数目和基因是否有功能去除。比如一个基因只有 *de novo * 注释结果来源支持,并且与公共数据库比对没有结果,可以考虑去除。
- 不同物种基因组的基因数目有差异,参考近源物种基因数目增删结果。
基因功能注释
虽然通过上面方法得到了基因组的基因序列,但是得到基因具体是什么基因,有什么功能,仍不清楚。这就需要进一步和一些公众数据库比对,确定基因功能。
注释思路如下:
- EggNOG 和 KEGG 都可以在线批量注释,就是得排队,比较慢;
- 自己本地下载数据最新文件,使用 diamond 构建索引,再比对就完事儿。
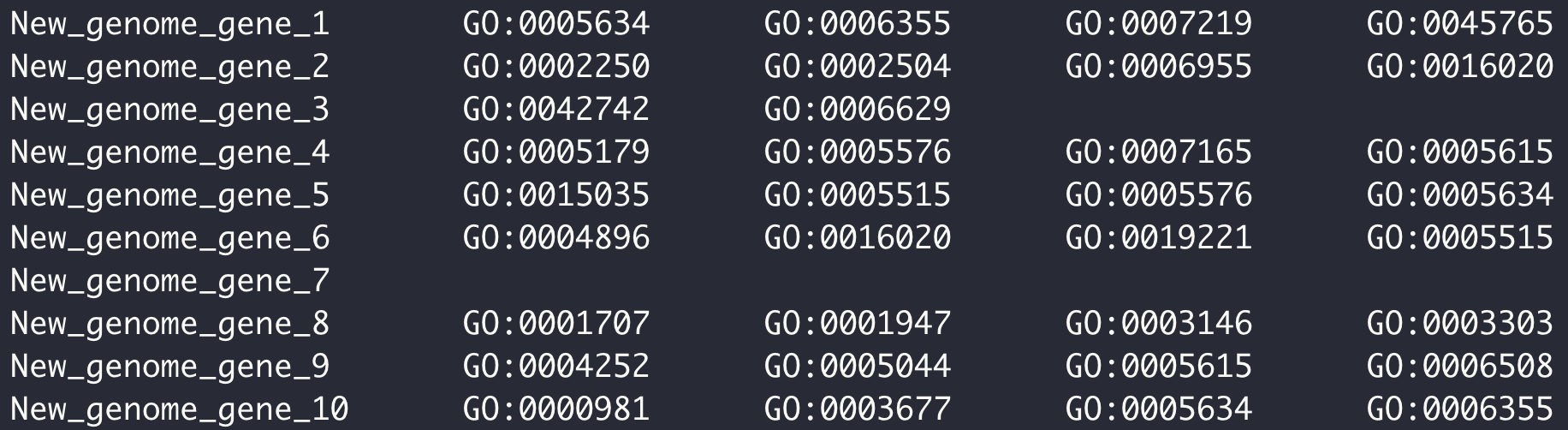
- KEGG 和 GO 富集分析背景文件制作,其实就是将基因 ID 和对应 pathway 或者 term 关联起来。以 OMA 数据库为例。
OMA 数据库中定义的序列 ID 各自有对应 GO term 号,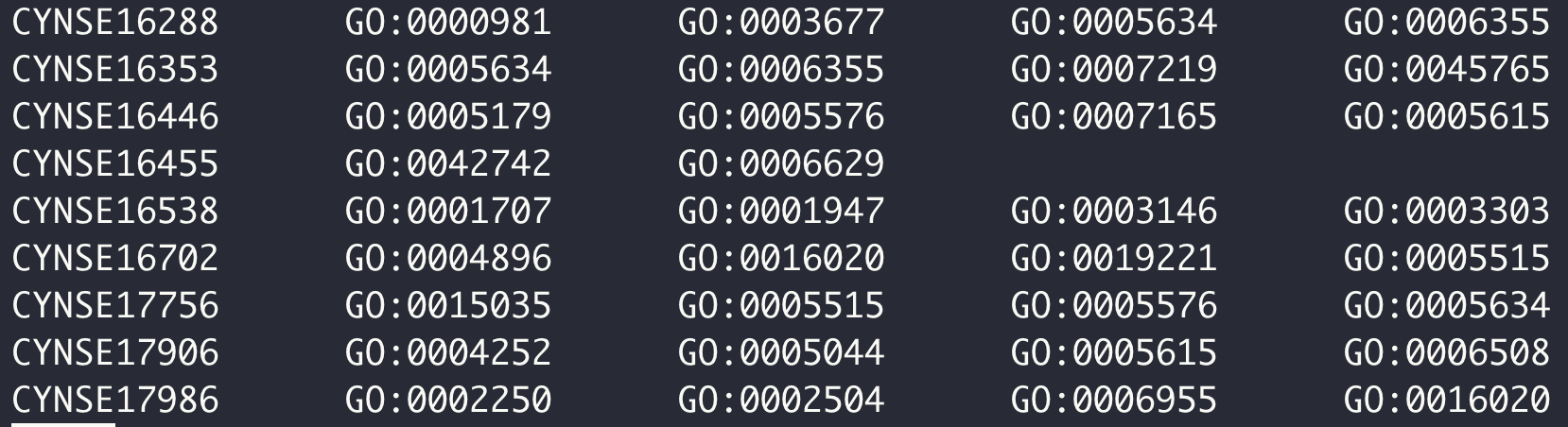
dimond 比对过滤后的结果有基因组基因 ID 和 OMA 数据库中定义的序列 ID 匹配的信息,
两个文件关联,替换 ID 名字,完事儿。