
从基因聊起(3)
在生信学习过程中,对基因展开分析,绕不开与gff文件打交道。
gff来源与格式介绍
GFF 全称Generic Feature Format,描述了基因组上各种特征的区间信息,包括染色体,基因,转录本等。
gff文件本质上是一个 “\t” (Tab 键) 分隔,共9列的纯文本文件。
关于 gff文件来源不一,导致gff文件格式有的千奇百怪,对于后续一些处理造成麻烦和可能影响分析结果(例如,一般进行基因家族分析时,根据gff文件先处理,选取最长转录本后再进行分析)。
不同的处理方式应对gff文件每一列信息内容有深刻的理解,才能更好进行相应的处理。
不同的 gff 格式介绍可去 Generic Model Organism Database 查看。
此处进行一些说明:平时使用较多的一种gff文件格式,其第三列只有mRNA和CDS信息,这里用一条mRNA信息代表一个基因信息,CDS指coding区,不可将其与exon在某些概念上混为一谈。
比如从 CNGB 上下载的gff文件会提供这样的格式。或者 Genewise 和 EVM 软件输出结果也是类似的格式(EVM 输出不考虑可变剪切,再对上文说的“用一条mRNA信息代表一个基因信息”解释)。
这种gff格式只是为了方便脚本提取、统计蛋白编码基因相关操作使用一种非标准gff2/gff3格式。示例如下: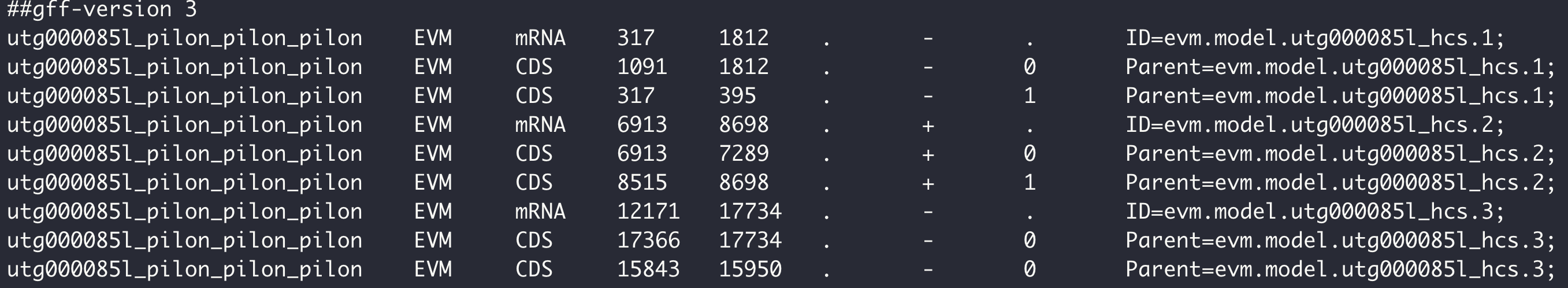 这种gff格式第9列 attributes信息进行了简化,只保留了基因的ID信息,要理解mRNA的“ID”与CDS的“Parent”之间相互关系。
这种gff格式第9列 attributes信息进行了简化,只保留了基因的ID信息,要理解mRNA的“ID”与CDS的“Parent”之间相互关系。
很多软件处理 gff 文件其实都与第九列的信息有关,要了解软件处理时真正抓取的是哪一块的信息。了解清楚之后,才能有效处理一些报错信息,又或者自己构造一些符合软件输入格式的文件
理解基因结构,理解 gff 文件注释信息。这里引用《组学时代》的一篇推文介绍。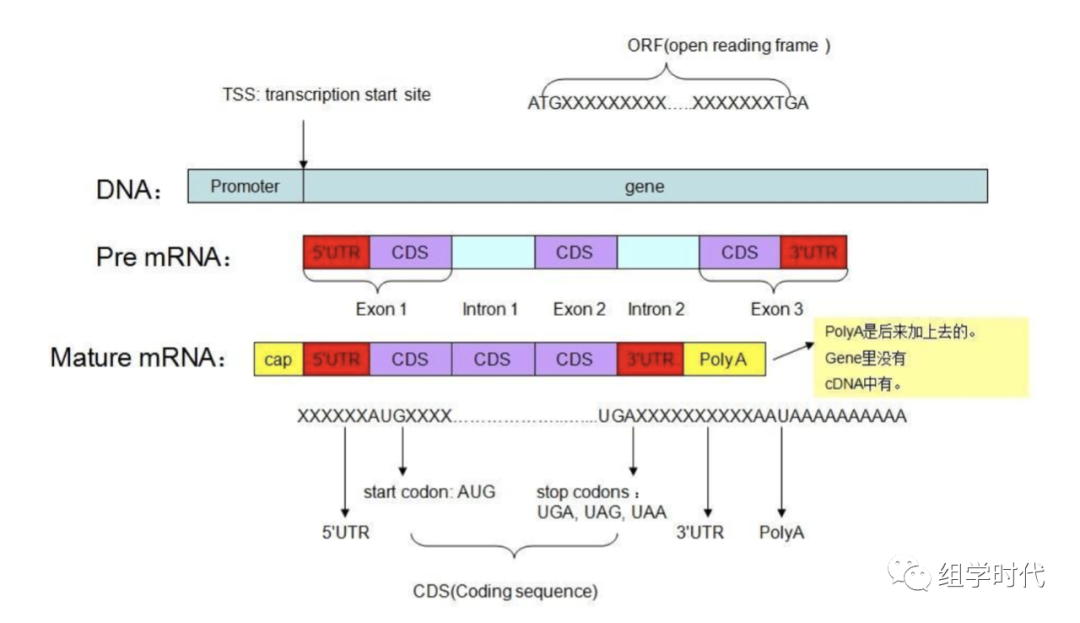
gff 文件处理通用流程
gff 文件统一格式
从一些格式不同的gff 文件只提取第三列为mRNA和CDS的信息,与对应的mRNA的“ID”信息,和CDS的“Parent”信息,统一格式,便于后续处理。
如pasa流程输出的pasa.gff处理为pasa.deal.gff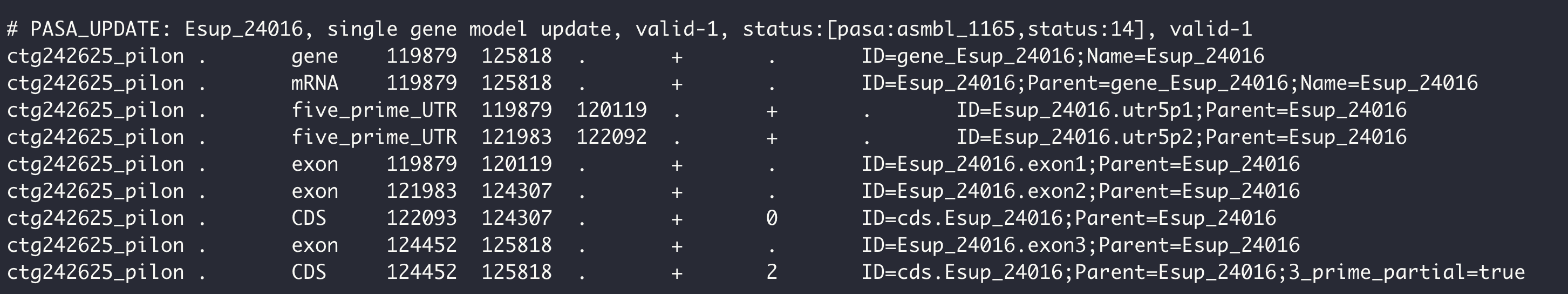
1 | awk '$3=="mRNA"||$3=="CDS"' pasa.gff | awk -F "[\t;]" '{if ($3=="mRNA") print $0"\t"$9";";else print $0"\t"$10";"}' | cut -f 1-8,10 > pasa.deal.gff |
但是这样的抓取会造成mRNA的长度和CDS长度不匹配的情况,如下: 可以使用fix_mRNA_coordinate.pl 进一步处理。
可以使用fix_mRNA_coordinate.pl 进一步处理。
1 | perl fix_mRNA_coordinate.pl pasa.deal.gff pasa.deal.fix.gff |
gff 格式转换
对于这样非标准的格式gff也可以使用 agat 工具转成标准gff3格式或者gtf格式。 gffread 软件也可以进行这样的格式转换和提供序列提取功能。
gffread 软件也可以进行这样的格式转换和提供序列提取功能。
提取最长转录本
有些分析需要我们对应提取最长转录本进行分析,有如下几种方法:
- 针对NCBI来源的gff
1
2perl pick_longest_ncbi.pl xxx.gff
### 自动在当前目录下生成名为clean.gff的文件 - 针对ensemble 来源的gff对于如上来源的gff,也有推荐使用 GetTransTool 工具处理。
1
2perl pick_longest_ensembl.pl xxx.gff
### 自动在当前目录下生成名为clean.gff的文件
- 其他来源的gff
有的可能就没有可变剪切的信息,这时候可以使用gffread软件check一下。如果第三列“locus”数目和“mRNA”数目相等,则此gff判断为无可变剪切信息。1
gffread -C -G -K -Q -Y -M -d dup xxx.gff > xxx.gffread.gff
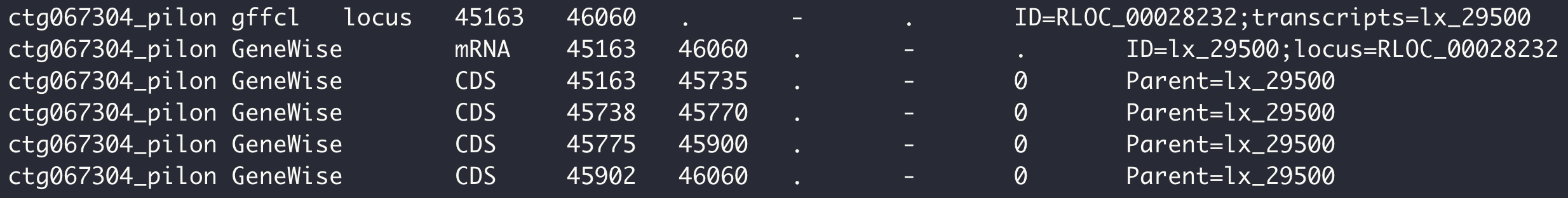 如果第三列“locus”数目和“mRNA”数目不等,可能存在可变剪切信息。可以先使用 agat 工具转换格式,再使用李恒大神新写的gffio软件提取最长转录本。
如果第三列“locus”数目和“mRNA”数目不等,可能存在可变剪切信息。可以先使用 agat 工具转换格式,再使用李恒大神新写的gffio软件提取最长转录本。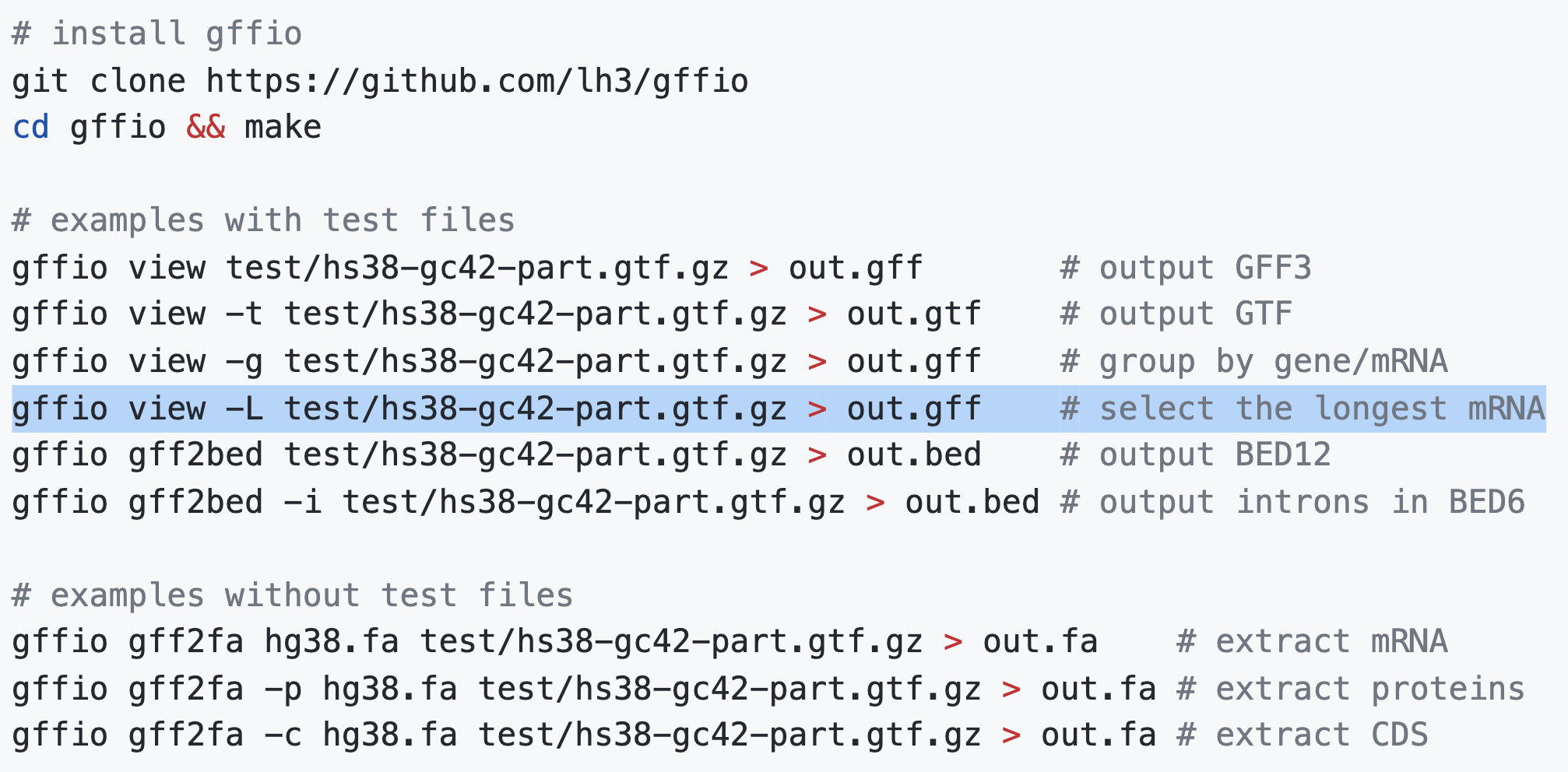 也可以先提取转录本序列,比较长短后,选择最长。PS:一般指转录本序列长度不是 mRNA 那一行的坐标直接相减。
也可以先提取转录本序列,比较长短后,选择最长。PS:一般指转录本序列长度不是 mRNA 那一行的坐标直接相减。 - 其他
对于高等脊椎动物的免疫相关基因存在重组等情况(如免疫球蛋白V(D)J基因重组),直接取最长转录本可能不是一个好办法,分析时需要都单独抓出来。
本文相关脚本下载链接:https://cowtransfer.com/s/cdefaa52d45e46
- 声明 fix_mRNA_coordinate.pl 为华大基因研究员孙帅(sunshuai@genomics.cn)所写
