
从基因聊起(1)
序列准确性
随着测序技术快速发展,大量的物种基因组、转录组等被测序。对这些物种的序列信息进行分析,有助于我们更多地认识生物演化,预测、了解基因的功能。
但是能准确对基因进行分析的前提是:这个基因就是我们要研究的那个基因——基因序列要准确。
再次确认你分析的基因
有些实验室的同学们可能就根据所看的文献中的提到的基因,也想在自己研究的物种中做做看。于是迫不及待地从数据库中找到该物种的基因直接就开始设计引物,准备大展身手。
别急,再确认一下你要研究的基因。
基因名注释错误
举个例子,ensembl 数据库中编号为 ENSORLG00000017796 基因注释为tlr7,但其实是 frmpd4 基因。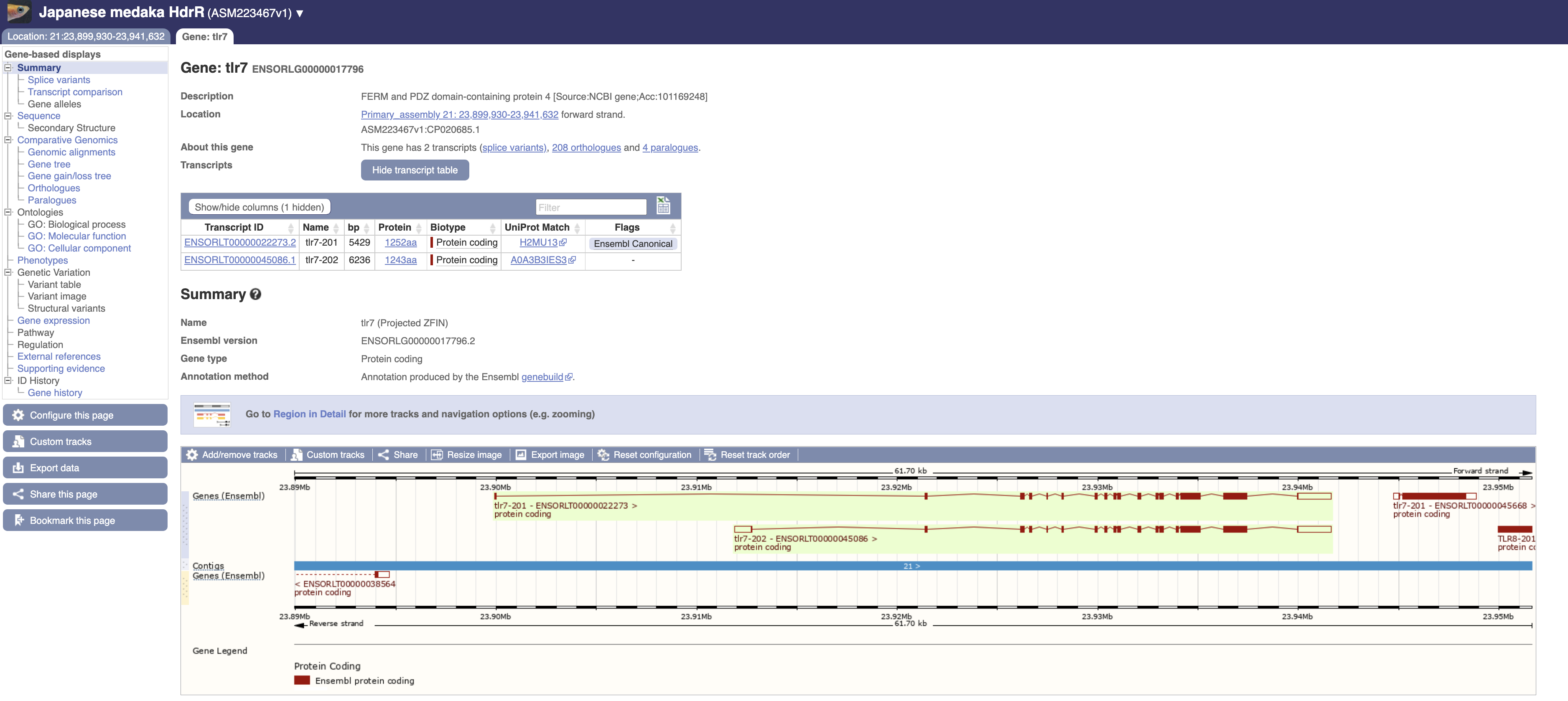
再次确认其结构域,也是与frmpd4相关。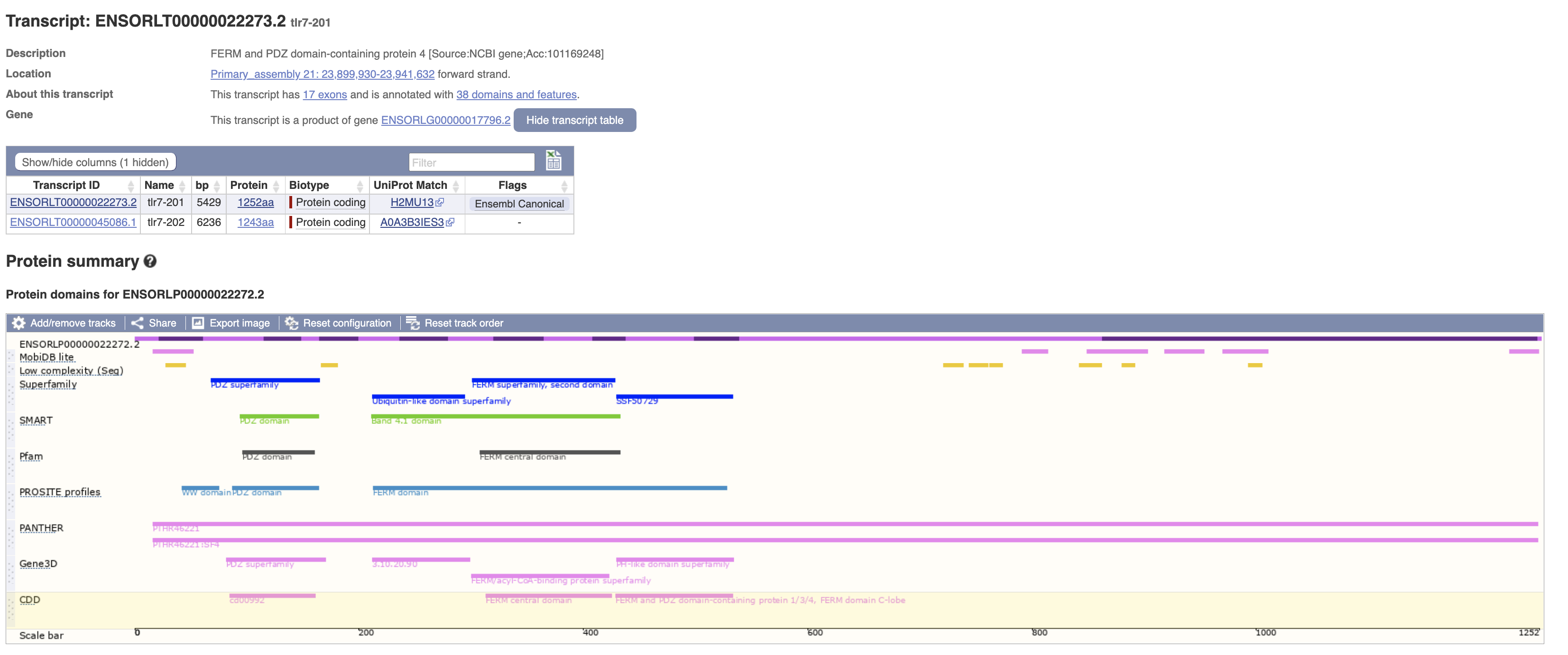
这个基因在NCBI 数据库中已经更新修正,但是 ensemble 数据库中还没更新修改。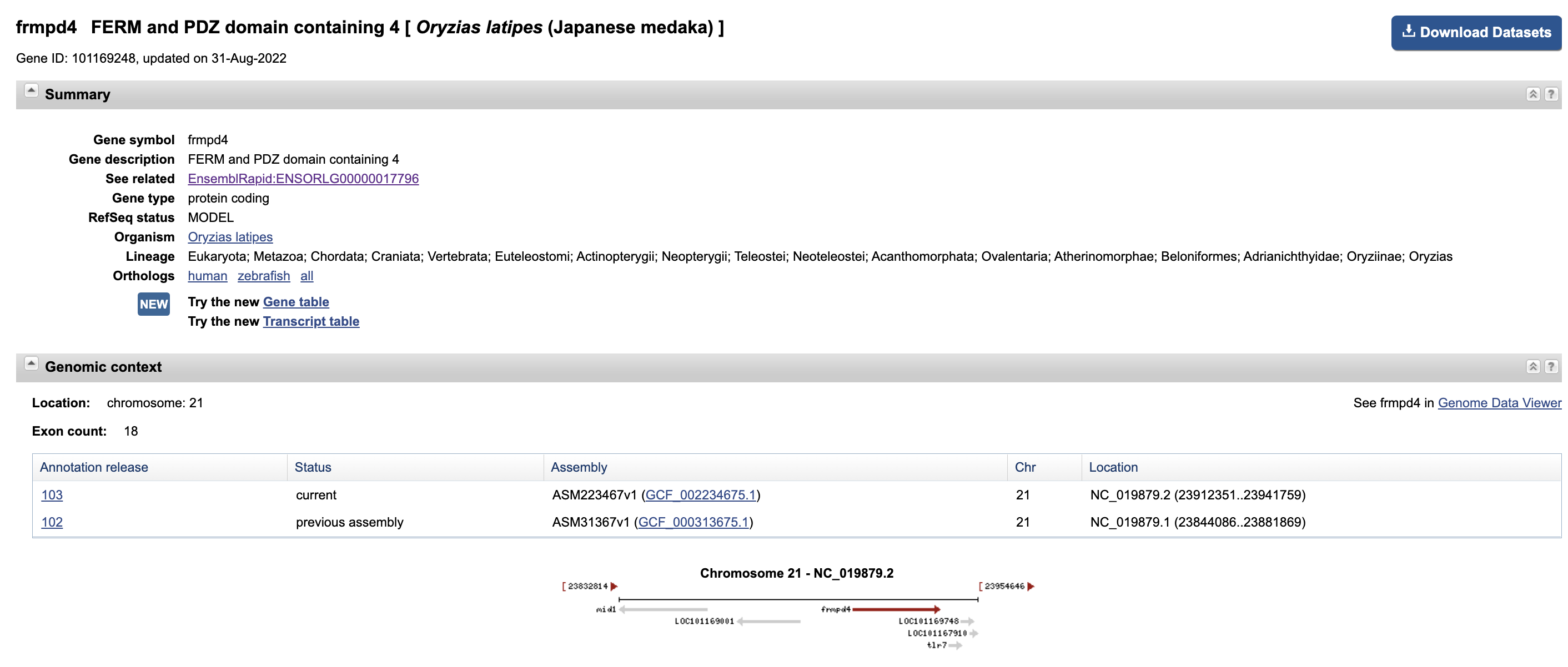
举这个例子是想说明:数据库中也有错误,需要多平台查询,论证。
选取代表性转录本
此处引用 CJ-chen 在其公众号一篇推文中的说法 (https://mp.weixin.qq.com/s/JIe-msPzBdJAAfcQlRhdcw) 
但是,是否选择最长转录本就“万事大吉”?
我觉得,可能得打个问号。
因为基因的表达是存在特异性的。
- 可变剪接是一种在转录后RNA水平调控基因表达的重要机制。一个基因通过可变剪接产生多个转录异构体,各个不同的转录异构体编码结构和功能不同的蛋白质,它们分别在细胞/个体分化发育不同阶段,在不同的组织,有各自特异的表达和功能。—— 引用来源 (https://www.plob.org/article/2730.html)
举这个例子是想说明:若分析的基因有多种预测的可变剪切类型时,确定选择“合适的”转录本进行研究分析。如何才算是合适,可能就只能自行在实验或者分析中摸索了。
多拷贝和假基因
这也是很令人头疼的情况,尤其是在数据公开较早的非模式生物中。
如果基因组组装质量较低的话,这些基因到底是真实的“多拷贝”还是“假基因”?如何确定?选哪条序列克隆、分析?实验结果怎样看……
那就只能多调研文献,具体情况具体分析了。
附上一些参考资料:
- 基因和转录本种类:https://vega.archive.ensembl.org/info/about/gene_and_transcript_types.html
- Wang M T, Li Z, Ding M, et al. Two duplicated gsdf homeologs cooperatively regulate male differentiation by inhibiting cyp19a1a transcription in a hexaploid fish[J]. PLoS Genetics, 2022, 18(6): e1010288.
- Lindsay M A, Griffiths-Jones S, Pink R C, et al. Pseudogenes as regulators of biological function[J]. Essays in biochemistry, 2013, 54: 103-112.
- 刘慧, 邹枨, 林凤. 假基因鉴定及其功能分析[J]. 生物工程学报, 2013 (5): 551-567.

评论
WalineTwikoo